Webpage update
May, 2026
Performing a parallel real-time TDSCF calculation of excitation energies and UV/Vis electronic absorption spectra (EAS) is a simple two-step process
respect --scf --inp=my-input-file --nt=4 --scratch=/path/to/scratch/directory respect --tdscf --inp=my-input-file --nt=4 --scratch=/path/to/scratch/directory
The first call of respect with the argument --scf performs the traditional time-independent self-consistent field (SCF) optimization of ground-state molecular orbitals, which is a necessary prerequisite for the following real-time TDDFT or TDHF calculation. The second call of respect with the argument --tdscf actually performs the EAS simulation using real-time electron dynamics.
The other arguments mandatory to respect are
--inp
specifies a name of the input file;
--nt
specifies the number of physical cores required for parallel execution;
--scratch
specifies a path to the scratch directory.
The input file my-input-file.inp should contain the input block scf: with some SCF-specific keywords and the input block tdscf: with some TDSCF-specific keywords. A comprehensive list of all keywords for SCF can be found here and for TDSCF here. A simple example of my-input-file.inp looks like
#non-relativistic 1c Hartree-Fock calculation of H2O molecule
scf:
geometry:
O 0.000000 0.000000 0.000000
H 0.000000 0.000000 0.940000
H 0.903870 0.000000 -0.258105
method: hf
basis: ucc-pvdz
charge: 0
multiplicity: 1
nc-model: point
maxiterations: 30
convergence: 1.0e-7
#RT-TDSCF calculation with a delta-type external field
#of amplitude 0.001 [a.u.] and x-direction. The real-time
#simulation will run for 2000 time steps of length 0.5 [a.u.]
tdscf:
spectroscopy: eas
time-steps: 2000 x 0.5
field:
model: delta
amplitude: 0.001
direction: 1.0 0.0 0.0
There are several important aspects associated with the RT-TDSCF simulations, namely
it is necessary to perform three independent RT-TDSCF simulations with the external field oriented along three perpendicular Cartesian directions
The easiest way to accomplish this requirement is to set the keyword direction: in the input block tdscf: to an unit vector along the x-axis as
direction: 1.0 0.0 0.0
or to an unit vector along the y-axis as
direction: 0.0 1.0 0.0
or to an unit vector along the z-axis as
direction: 0.0 0.0 1.0
and rerun the RT-TDSCF simulation for each direction. Importantly, save the TDSCF output files with specific suffixes -- _x.out_tdscf for x-direction, _y.out_tdscf for y-direction, and _z.out_tdscf for z-direction. These name-specific files are required for a final post-processing of the time-domain signal and visualisation of spectra (see next point).
final post-processing of time-domain signals and visualisation of spectra using the script spectrum.py
After having the three TDSCF outputs files, named for instance as my-output-file_x.out_tdscf, my-output-file_y.out_tdscf, and my-output-file_z.out_tdscf, the final electronic absorption spectrum (EAS) can be obtained by running
/path/to/ReSpect/util/spectrum.py --files=my-output-file --spectroscopy=eas --plot --smooth
For the example above, the EAS spectrum looks like
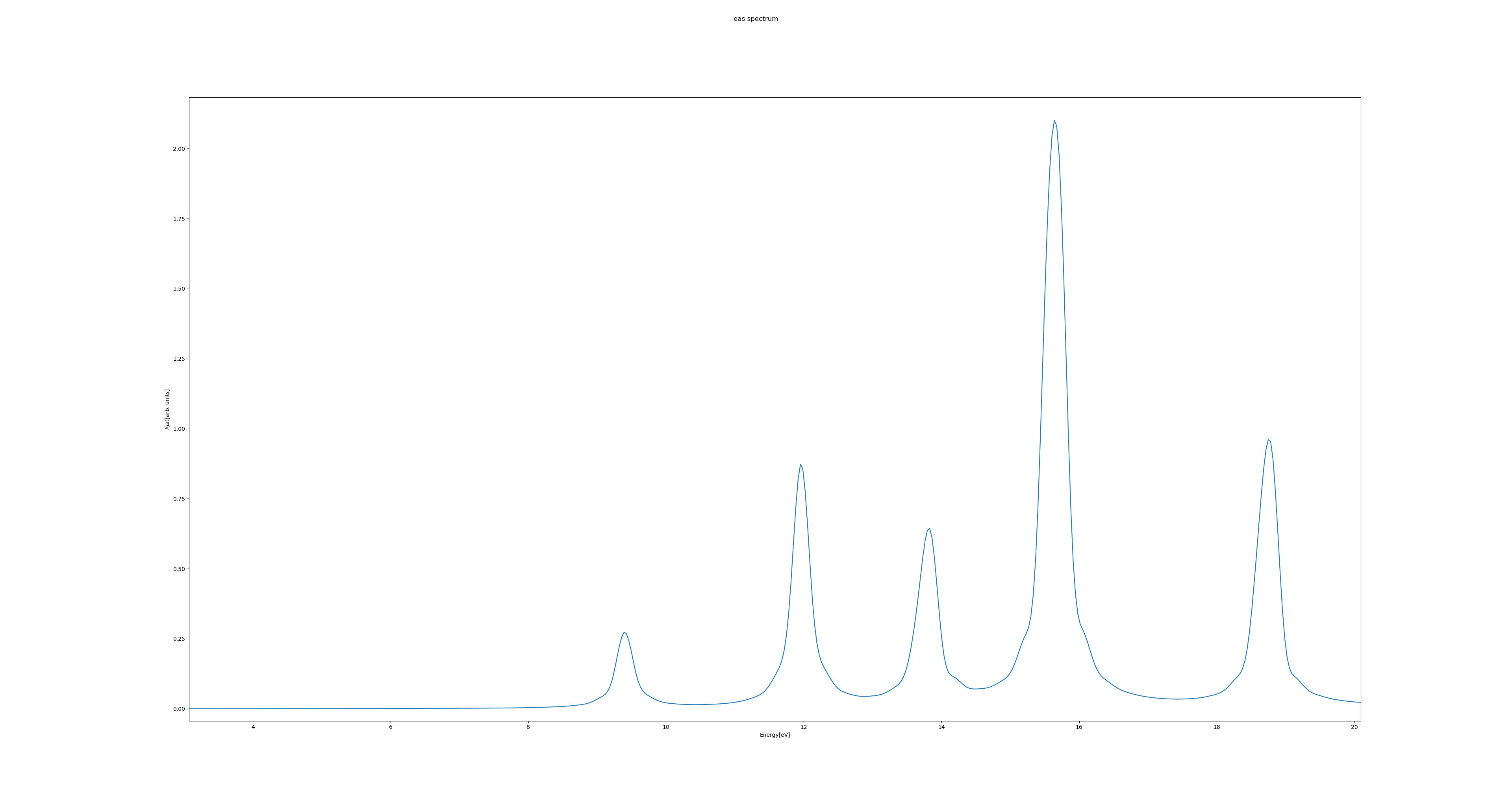
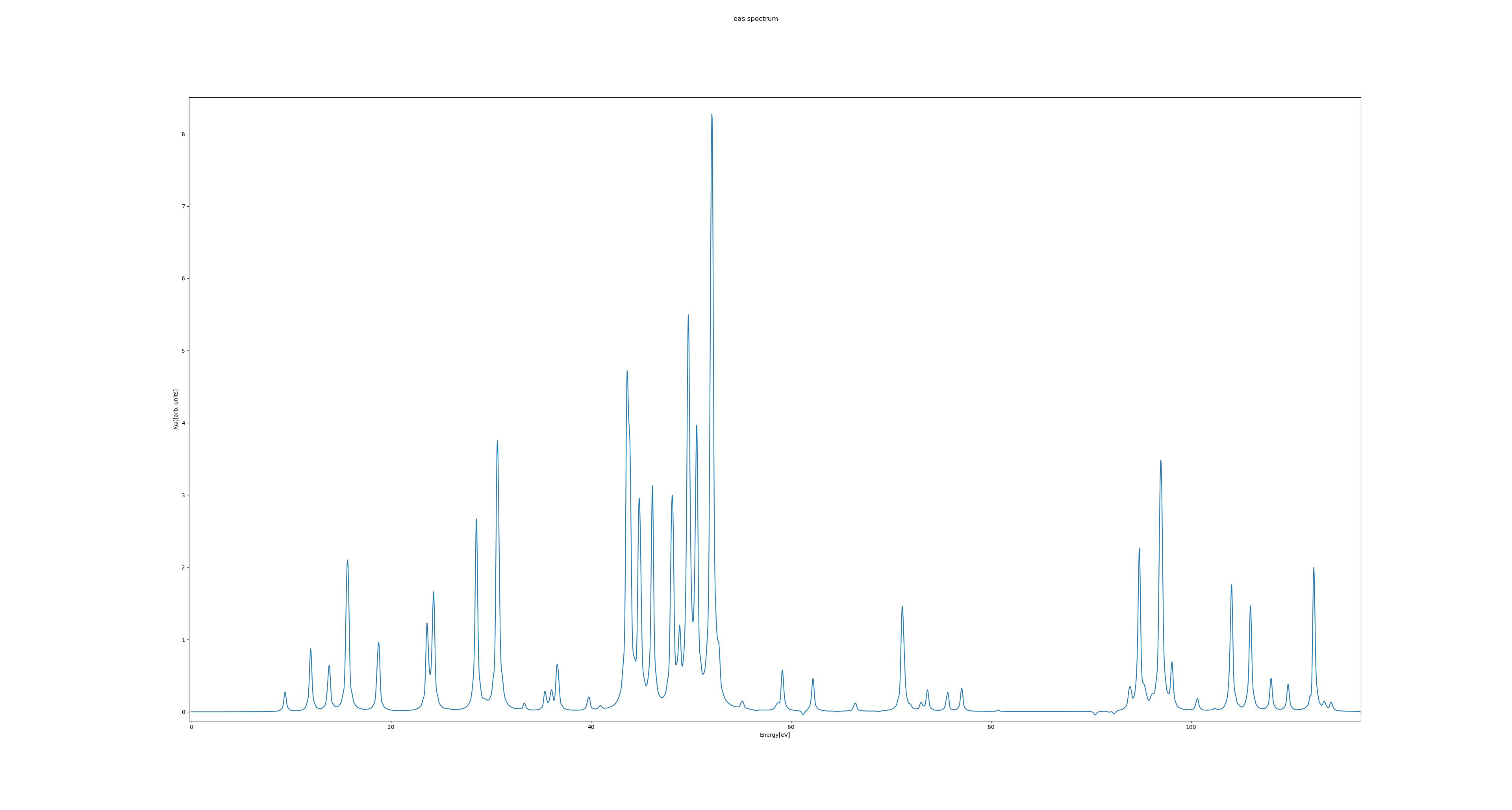 Of relevance for UV/Vis spectroscopy is the energy range between approximately 1.4 eV and 12.4 eV, corresponding to wavelengths of about 900 nm to 100 nm.
Of relevance for UV/Vis spectroscopy is the energy range between approximately 1.4 eV and 12.4 eV, corresponding to wavelengths of about 900 nm to 100 nm.
the script spectrum.py will also generate two files; my-output-file.out_spectrum_eas containing all datapoints of the frequency domain spectrum and my-output-file.out_peaks_eas containing datapoints above the threshold 10-7 and information about the post-processing settings, relevant physical constants, and spectral resolution. All arguments to the script spectrum.py can be listed by the command
/path/to/ReSpect/util/spectrum.py --help
the frequency-domain results are obtained by discrete Fourier transform of the time-domain results. Therefore, a time-domain simulation that consists of \(N\) time steps each of length \(\Delta t\), the Fourier transform yields the frequency-domain result with a resolution \(\Delta\omega\) within a finite frequency interval of length \(\Omega\) $$ \Delta\omega = \frac{\Omega}{N} \qquad \Omega = \frac{2\pi}{\Delta t} $$ For the example above, \(N=2000\), \(\Delta t=0.5\)au, \(\Delta\omega=0.17\)eV, and \(\Omega=342\)eV. The previous expressions tell us that in order to increase the resolution in calculated spectra we need to increase the total simulation length \(N\Delta t\) by increasing the number of time steps (which makes the simulation more time consuming) and/or increasing the size of the time step (which puts extra demands on the propagation solver). In addition, because the frequency interval \(\Omega\) depends inversely on the time-step length, in order to describe high-frequency parts of spectra, such as in X-ray spectroscopies, shorter time steps are required. Therefore, a balance between the resolution, frequency range and computational cost has to be achieved by choosing suitable simulation parameters.
There are several important and worth-to-remember aspects associated with the input syntax, namely
the input is case-insensitive
This means that the program does not distinguish between uppercase and lowercase letters.
the input is insensitive to the number of blank lines and/or comment lines
All comments begin with the number sign (#), can start anywhere on a line and continue until the end of the line.
the input is compliant with the dictionary syntax of the YAML markup language
This means that each input line is represented either by a single block: statement or by a simple keyword:value pair, such as
block1:
keyword1: value1
keyword2: value2
...
block2:
keyword3: value3
keyword4: value4
...
block3:
keyword5: value5
keyword6: value6
...
It is essential to remember that all members of one block: are lines beginning at the same indentation level. Whitespace indentation is used to denote the block structure; however, tab characters are never allowed as indentation. The only exception to the YAML-based input syntax is the block geometry: which utilizes a simple xyz format for the molecular geometry specification.
Q: How to scale the speed of light in the RT-TDSCF calculation?
Set the cscale option in the SCF calculation. The scaling value is then automatically transferred to the RT-TDSCF calculation.
Q: Is it possible to scale spin-orbit interaction in the RT-TDSCF calculation?
No. Currently this feature is not supported.
Q: Is there a way to launch RT-TDSCF calculations without the need to explicitly setup the scratch path by "--scratch=/path/to/scratch/directory"?
Yes, the argument "--scratch=/path/to/scratch/directory" can be saved to the file .respectrc in your home directory. If both the file and the command line argument exist, then ReSpect takes the scratch directory setting from the command line.
Q: How to restart a RT-TDSCF calculation?
RT-TDSCF calculations can be restarted simply by creating the symbolic link to the data file of the previous job
ln -sf my-old-job.50 my-new-job.50
and by adding an extra argument --restart to the command line when restarting new job
respect --tdscf --inp=my-new-job --nt=4 --scratch=/path/to/scratch/directory --restart
The restart process collects and prints to the output file my-new-job.out_tdscf all relevant RT-TDSCF data generated in the previous run.
Q: Is it possible to recover a RT-TDSCF job from a scratch directory?
If the ReSpect job for any reason fails and the scratch directory is still accessible, it is possible to recover the failed calculation. After running the command
respect --recover-job=/path/to/scratch/directory
file respect_checkpoint_file.50 will be created in the current directory. Using this file, you can now restart the RT-TDSCF calculation as described in the tip How to restart a RT-TDSCF calculation?.